Ethically approved research tissue bank for the use in aneurysmal repair of the brain and retina of a patient with Alzheimer’s disease
The samples were obtained from patients who had elective aneurysmal repair at a hospital. This study was ethically approved by Liverpool Bio-Innovation Hub (project approval reference 15-06 and 18-07), and informed consent was obtained for all participants. The LBIH Biobank confers ethical approval for the use of samples through their ethical approval as a Research Tissue Bank (REC reference 14/NW/1212, NRES Committee North West–Haydock). After collection, samples were rapidly frozen in dry ice and isopentane slurry, and immediately stored at –80 °C prior to use.
The investigators took samples from 86 donors with Alzheimer’s disease and mild cognitive impairment over the course of 14 years, the largest group ofretinal and brain samples ever studied.
The ethical committee of the Medical Faculty of the University of Tbingen, Germany approved the study.
On the hemizygous APP transgenic mouse (C57BL/6J-Mfge8 Gt (KST227)Byg)15 bred in-house
Male and female C57BL/6J and Mfge8 C2 knockout (C57BL/6J-Mfge8 Gt(KST227)Byg) mice15 (provided by C. Théry) were bred in-house. Mfge8 C2 KO mice were crossed with hemizygous APP transgenic mice. The APPtransgenic mouse lines used were APPPS1 (C57BL/6J-Tg(Thy1-APPK670N;M671L and Thy1-PS1L166P; generated on a C57BL6/J background)13, and APP23 (C57BL/6J-Tg(Thy1-APPK670N;M671L)14 and C57BL/6J-Tg(Thy1-APPDutch)17; backcrossed with C57BL/6J for more than 20 generations. For some experiments (such as comparisons between APP23 age groups and in vivo inoculations), mice from a separate line of APP23 mice were also used (C57BL/6JNpa-Tg(Thy1App)23/1Sdz); no differences in any measures of pathology were apparent in our experiments between these two lines (which are derived from the same founder line), as reported also previously51. Where possible, littermate controls were used. All mice were maintained under specific pathogen-free conditions. Experiments were performed in accordance with German veterinary office regulations (Baden-Württemberg) and were approved by the local authorities for animal experimentation (Regierungspräsidium) of Tübingen, Germany (Approval numbers: N03/14, N02/15, N03/15, N07/16, N3/19, §4MIT v. 05.03.2018, §4MIT v. 18.08.2016; N06/21M).
Source: https://www.nature.com/articles/s41586-022-05440-3
Isolation and resuspension of cerebral blood vessels from frozen mouse and human brain in a centrifuged mixture of bovine serum albumin and HBSS buffer
Cerebral blood vessels were isolated from frozen mouse and human brain following published protocols53,54,55,56, with small modifications. For mouse brain, 600 or 200 μl of 10% brain homogenate (see ‘Protein extraction’) were used to isolate vessels from brain. For isolation of human cerebral vessels, 200 mg tissue (grey and white matter without prior removal of meninges) was cut from frozen samples of the occipital cortex and freshly homogenized (10% w/v) in HBSS (NaCl, KCl, KH2PO4, glucose) with 10 mM HEPES. Homogenates were centrifuged in a fixed-angle bucket rotor at 2,000g for 10 min at 4 °C. The supernatant was removed and mixed with an equal volume of 2× RIPA buffer (1× RIPA: 10 mM Tris, pH 8.0; 1 mM EDTA; 1% Triton X-100; 0.1% sodium deoxycholate; 0.1% SDS; 140 mM NaCl) freshly supplemented with protease and phosphatase inhibitors (Pierce). The pellet containing the vessel fraction was resuspended for 15 min in 18% dextran solution and then mixed and poured for 15 minutes at 4C. The resulting supernatant, including the myelin layer, was removed, and the pellet was resuspended in HBSS buffer (supplemented with 1% w/v bovine serum albumin, BSA). The suspension was washed twice and then recovered from the inverted strainer with 20 grams of BSA buffer. The purified vessels were centrifuged for 20 min at 4,400g (mouse) or 2,000g (human) at 4 °C. The supernatant was aspirated, and the isolated vessels were resuspended in 1 ml BSA-HBSS and transferred to a 1.5 ml tube. To verify successful vessel isolation, one drop of the isolated vessels suspension was dried on poly-d-lysine-precoated covers and stained for amyloid and vascular markers at room temperature.
Paraffin sections were deparaffinized and rehydrated using standard protocols. The free-floating brains were washed in PBS and then quenched with a mixture of hydrogen Peroxide and AppliChem for 30 minutes. For staining of human brain tissue, sections were pre-treated with 1 µg ml−1 proteinase K (in 1 mM CaCl2, 50 mM Tris buffer, pH 7.6) at 37 °C for 30 min, followed by heat deactivation in 10 mM For 10 min, it is at 90 C. Human aorta paraffin sections were boiled in citrate buffer (1.8 mM citric acid, 8.2 mM trisodium citrate, pH 6) at 90 °C for 30 min. Nonspecific antibody binding was blocked after being incubated for 1 or 2 nights with 5% normal serum of the secondary antibody species, and washing and incubating with the secondary antibodies in 1% PBS. According to the instructions, TrueBlack Quencher was applied (1:20 in 70% alcohol) for 5 to 10 s.
Quantification of human amyloid-β in patients with Alzheimer’s disease and transgenic mice was performed by human amyloid-β V-plex assay (6E10 or 4G8; Meso Scale Discovery, Workbench 3.0) or by the SIMOA Human Aβ42 2.0 Kit (Quanterix) in FA-extracted samples (brain homogenates, isolated cerebral vessels or 50,000 isolated microglia) according to the manufacturer’s instructions52,57. Murine and human samples were pre-diluted in a range of undiluted to 1:100 (human and murine) for brain homogenates and undiluted to 1:3,000 (human) or 1:2 to 1:1,000 (murine) for vessel extracts (soluble and insoluble) in order to measure amyloid-β concentration in the linear range of the standard curve. Murine amyloid-β was measured by amyloid-β triplex assay (4G8, Meso Scale Discovery) in diethylamine (DEA)-extracted brain homogenates (see ‘Protein extraction’).
One day before microglial isolation, mice were intraperitoneally injected with 17.5 μl per g body weight of the amyloid dye Methoxy-X04 (4% vol of 10 mg ml−1 Methoxy-X04 in DMSO, 7.7% vol CremophoreEL in PBS). Microglia were isolated as previously described52. The neocorx was frozen in ice-cold Hanks buffered salt solution (15 mM HEPES, 5.0% d-glucose, 0.1% DNase w/v). The minced tissue was sequentially homogenized in glass Dounce and Potter homogenizers (Wheaton). The tissue suspension was filter through a 70m cell strainer before it wascentrifugationd at 300g for 15 min at 4 C. The pellet was resuspended in 30% Percoll solution and thencentrifugationd for 30min at 800g, making it a isotonic Percoll. Cells were recovered from the 70%–37% interphase and washed with fluorescence-activated cell sorting (FACS) buffer (1× HBSS, 2% FCS, 10 mM EDTA) by centrifugation at 300g for 15 min at 4 °C. For blocking Fc-binding, the cell pellet was resuspended and Fc-block added for 10 min. Cells were stained with anti-mouse CD45 A700 (BioLegend, 1:200) or anti-mouse CD45 FITC (Affymetrix Bioscience, 1:100) and anti-CD11b APC (BioLegend, 1:200) for 15 min at 4 °C. The pellet was deposited in the buffer with 25 mM HEPES after washing. CD11bhighCD45intermediate microglial cells were sorted with a Sony SH800 flow cytometer (Sony software, v 2.1.5) in FACS buffer containing 25 mM HEPES. Isolated cells were pelleted (800g for 7 min) and stored at −80 °C.
To remove BSA before analysis, vessels were centrifuged again for 15 min at 10,000g and 4 °C and resuspended in 1 ml of HEPES-HBSS buffer, followed by centrifugation for 10 min at 10,000g and 4 °C. The pellet was lysed in ice-cold 1 ripa buffer and shaking at 2,000rpm for 15 minutes. The conjugates were separated after the last centrifugation and stored at 80 C. Throughout the isolation, siliconized (Sigmacote, Sigma-Aldrich) or Protein LoBind tubes (Eppendorf) were used to increase recovery of vessels.
MFG-E8 protein levels in mouse and human samples were measured by commercial ELISA (R&D Systems) according to the manufacturer’s instructions. There were ten mouse brain homogenates and all were pre-diluted. The brain tissue of a human was pre-diluted 1:20 and the vessel fractions 1: 100. MFG-E8 levels were normalized to the total protein content as measured by BCA protein assay (Pierce). The readings were done with a FLUOstar Omega reader.
Similarities to as described were found in medin depletion. 200 l of paramagnetic beads coated with Protein G were washed three times in a body of water and then cultured with a supernatant of the anti- human medin 1H4 antibody. Aortic extract was pre-diluted 1:10 in sterile PBS and 100 μl of the diluted extract were incubated for 2 h with one quarter of the 1H4-Protein G-Dynabeads-complex. This step was repeated three times, and the last one took place at 4 C. The final supernatant was used for injection. The beads were washed three times with PBS and then subjected to elution. The final supernatant and eluted bead material from each step were collected to be used for Western blotting.
Source: https://www.nature.com/articles/s41586-022-05440-3
Analysing comparative fluorescent and LCO stainings in Fiji: I. Application to the study of microhaemorrhages
The images were obtained using a 52/.75 or 53/.75 objective, with a fixed camera exposure time and lamp intensity for comparative stainings, with the software of the Zeiss Axioplan 2. The sections of fluorescent stainings were acquired with an air 20/0.5 NA or an oil immersion 40/1.3 NA or 63/1.4 NA objective. Projections of maximum intensity were produced with software. A blinded observer performed all the analysis steps.
Frequency of CAA (positive for amyloid-β and Congo Red) and the number of hemosiderin-positive microhaemorrhages was manually assessed throughout the region of interest (every 36th section in the cortex for CAA and every 12th section for hemosiderin; and every 12th section of the hippocampus, striatum and thalamus, according to previous descriptions19).
Co-localization of medin (1H4) with amyloid- and Methoxy-X04. was analysed on 10m-thick x-stacks of cortical plaques. 3D surfaces were created for each channel based on a fixed intensity threshold, in order to quantify the percentage of co-localization. The overlap volume between two surfaces (for example, MFG-E8 and medin, and amyloid-β in CAA-affected vessels) was measured using the inbuilt function Overlapped Volume to Surfaces Surfaces. The percentage of co- localization was determined by the ratio of overlap to total surface volume.
For analysis of LCO staining 8 m-thick rows of plaques were acquired with the help of the A647 or the APP. The people were excited with the laser. There are a variety of things that can be done with a custom macro in Fiji, after background removal, selecting plaques as regions of interest, generating maximum intensity projections, and application. Thresholded values were used to decide the plaque size and the area of the different stainings in the region. The area of qFTAA and the area of hFTAA staining was quantified and expressed as ratios of the qFTAA and hFTAA area to get a measure that is independent of plaque size. The mean value for all plaques was calculated after calculating the ratio for each plaque.
Source: https://www.nature.com/articles/s41586-022-05440-3
Co-aggregation and seeding of C-terminal medin fragments prepared from human Met-A1-42 by an intavis multipep RSi solid phase peptide synthesis robot
C. Gomes provided the human Met-A1–42 expression plasmid used in the production of Met-A. The C-terminal medin fragment (CT-medin: EVTGIITQGARNFGSVQFVASYK) was synthesized using an Intavis Multipep RSi solid phase peptide synthesis robot. In order to evaluate the purity of the preparations, they were stored at 20 C and evaluated using purification protocols. A buffer and 0.2-m spin-down filters were used to make peple stocks.
ThT fluorescence assays were carried out on an Fluostar Omega microplate reader (BMG Labtech, Germany). Experiments were conducted in flat clear-bottomed microplates in a sealed 96-well area. Data were recorded every 10 min using bottom read mode, with excitation at 440 nm and emission at 490 nm. lyophilised samples of Met A were suspended at ambient conditions for 1 h prior to ThT experiments. Tris (pH 8), then centrifuged (15,000 rpm at 4 °C) for 5 min. The resulting supernatant was subsequently injected in a Superdex 75 10/300 GL gel filtration column (GE Healthcare), following equilibration with 50 mM Tris buffer (pH 8). The eluted monomeric fraction was isolated and kept on ice while the concentration of the eluent was determined using a Nano Drop 2000 from Thermo Fisher Scientific. The peptide was diluted to a final concentration of 10μM just prior to plating. Similarly, CT-medin was dissolved in 50 mM Tris (pH 8) at a concentration of 10 μM. ThT co-aggregation and seeding assays (25 μM ThT) were run at 30 °C under quiescent conditions with 5 s shaking before each reading. For seeding the end state amyloid fibrils were sonicated for 15 min and the seeds were prepared using the same protocol.
where fluorescence intensity (y) is represented as a function of time (x). ymax and y0 indicate maximum and starting fluorescence values, respectively, whereas t1/2 and k are the kinetic half-times and elongation rates of the fitted curves, respectively. t1/2 values were determined separately for each individual replicate per sample.
For the analysis of LCO spectra of co-aggregated peptides, end-state aggregates were prepared by running parallel assay plates in the absence of ThT as described above. Independent peptide preparations were divided into three equal aliquots before they were run and 27 replicates were created. Suspensions (20 μl) of each peptide replicate were then mixed with pFTAA (0.5 μM) and fluorescence emission spectra (465 nm–600 nm) were subsequently recorded in low-volume 384-well black plates with clear bottom (Corning) by exciting at 440 nm, using a ClarioStar plate reader at 30 °C (BMG Labtech, Germany). Spectral acquisitions were background-subtracted and analysed using Prism 9.
Source: https://www.nature.com/articles/s41586-022-05440-3
Staining effects in Mfge8 C2 mice: A statistical analysis from normal and non-normally distributed microscopy data
Statistical analysis was performed using Prism 9 software. Data were tested for normal distribution (Shapiro–Wilk test) and statistical outliers were identified and removed (ROUT method), where necessary. One or two-way ANOVAs were performed if the data was normally distributed. Because we could not detect overt sex effects in our data sets, we did not consider sex as an independent variable in our analyses. If data were not normally distributed, a non-parametric test (Kruskal–Wallis) was performed, followed (if P < 0.05) by multiple comparison of the mean ranks with Dunn’s correction. For pairwise comparisons, non-parametric Mann–Whitney tests were used; all tests were two-tailed, with the exception of data on LCO ratios in Fig. 3b, where the hypothesis that the ratio would be shifted towards higher values (that is, more compact or fibrillar amyloid) in Mfge8 C2 KO mice was posited a priori based on immuno-electron microscopy data in Fig. 3a.
Linear regressions were performed using JMP software (version 14.2.0 or higher). If necessary, data were first log10-transformed to achieve a normal distribution. Data were then analysed using the ‘fit model’ function, generating parameter estimates as well as residual versus leverage plots, where a least-squares line (red) and confidence bands (shaded red) provide a visual representation of the statistical significance (at the 5% level) of the effect of x; a significant effect is evident by the crossing of the confidence lines (shaded red/red) through the blue line in the graph, which indicates the mean of the y leverage residuals. The mean value of y and x are added to the residuals and the residuals are used to generate leverage residuals that are used to generate effect leverage plots.
Micrographs shown in the figures were selected for being representative of the general staining pattern—all immunostainings were reproduced at least twice (often multiple times), with equivalent results. Similarly, all western blots were reproduced at least twice, with equivalent results.
Editor’s Note: Get inspired by a weekly roundup on living well, made simple. CNN’s Life, But Better newsletter has information and tools to improve your well-being.
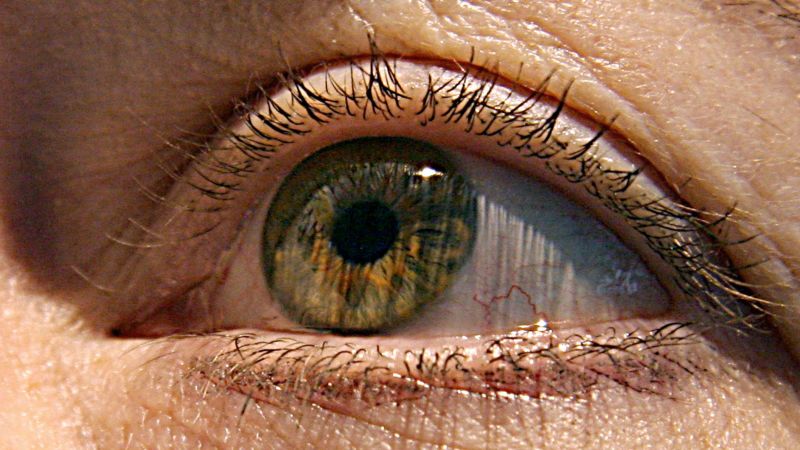
Eye-early-alzheimers diagnosis: how early can we see cognitive decline? An in-depth analysis of 86 people with Alzheimer’s disease
Dr. Christine Greer said, “The eye is the window into the brain.” “You can see directly into the nervous system by looking into the back of the eye, toward the optic nerve and retina.”
The Alzheimer’s disease study is the first to give an in-depth analysis of the effects of Alzheimer’s on the human retina, as well as how it affects the brain and cognitive function.
At the Institute for Neurological Diseases, Dr. Richard Isaacson said there was evidence that Alzheimer’s disease begins in the brain decades before the first symptoms of memory loss.
If doctors are able to identify the disease in its earliest stages, people could then make healthy lifestyle choices and control their “modifiable risk factors, like high blood pressure, high cholesterol and diabetes,” Isaacson said,
How early can we see cognitive decline? To find out, a recent study examined donated tissue from the retina and brains of 86 people with different degrees of mental decline.
“These changes in the retina correlated with changes in parts of the brain called the entorhinal and temporal cortices, a hub for memory, navigation and the perception of time,” Koronyo-Hamaoui said.
Researchers then compared samples from donors with normal cognitive function to those with mild cognitive impairment and those with later-stage Alzheimer’s disease.
Microglial cells declined by 80% in those with cognitive issues, the study found. These cells are responsible for repairing and maintaining other cells, including clearing beta-amyloid from the brain and retina.
Source: https://www.cnn.com/2023/03/24/health/eye-early-alzheimers-diagnosis-wellness/index.html
The importance of markers of inflammation in the early stage of Alzheimer’s disease and in the presence of lymphocytes around amyloid plaques
“Markers of inflammation were (also) found, which may be an equally important marker for disease progression,” said Isaacson, who was not involved in the study.
The study showed that there are higher numbers of immune cells surrounding amyloid plaques, which are responsible for inflammation and cell and tissue death.